
VALIDATION
Clean Room /HVAC Validation
- QACAL follows a quality management system (QMS) which includes standard operating procedures (SOPs), processes and data management, customer feedback and man-material movement with training and traceability records to ensure that we are holding ourselves to the highest level of quality.
- QACAL follow regulatory guidelines such as ISO 14644, cGMP, cGLP, cGxP, cGDP, GAMP 5, 21 CFR Part 11, 21 CFR Part 210 as well as 21 CFR Part 211.
- The revised versions of ISO 14644 Parts 1 and 2 introduce changes to sampling procedures and monitoring plans for cleanrooms and clean zones.
- The ISO14644-1& 2 provides a total validation approach related to Cleanrooms and associated controlled environments ISO 14644-1 annexure B provides test methods for designated classification of airborne particulate cleanliness and for characterizing the performance of cleanrooms and clean zones.
- ISO 14644-3 is consisting of the following tests as part of HVAC/Cleanroom validation.

CIP/SIP VALIDATION SERVICES
- SIP is a widely adopted method for the inline sterilization of processing equipment.
- The main advantage of SIP is the reduction of aseptic connections and manipulations that might compromise the integrity of the downstream equipment.
- Likely to be applied more and more with isolator and RABS technology.
- ISO 13408-5:2006Aseptic processing of health care products — Part 5: Sterilization in place
- SIP (Sterilize, or Steam In Place) is a timed sterilization of the upstream and downstream pharmaceutical production line using clean steam. It is part of a 5-step sanitization routine that occurs after every production batch, and follows the final rinse after CIP (Clean In Place).
- SIP ensures that every square inch of the production line that comes in contact with drug substance inputs, drug substance, or the final drug product is “sterilized” to ensure that there is no microbiological activity in the system. Clean Steam (made from USP Purified Water) is circulated through all of the process tubing during this stage, and enters large vessels through spray balls embedded in the vessel ceiling.
- SIP is a temperature validated process, meaning that the sterilization event must be proven by measuring the temperature of the event and recording the data. The minimum sterilization regimen requires the injection of clean steam into all piping and vessels for at least 1/2 hour after they reach a minimum temperature of 250°F (121°C). If the temperature ever falls below 250°F (121°C) during the temperature hold period, a temperature validation fault is recorded, and SIP must be repeated.
- Validation temperature sensors (usually thermocouple) are placed at the condensate outlets of process equipment to make sure that the sterilization temperature meets the specific regimen designed for the process system. The sensing elements are usually designed with integral sheathes and Tri-Clamp connections and are clamped directly to tubing tees, or the element is inserted into a Tri-Clamp thermowell connected to the tee. The sensors are normally located twelve to eighteen (12 – 18) inches (300 – 450mm) upstream of the clean steam trap where the condensate exits the piping or vessel


- QACALprovides Turnkey Advanced Thermal validation services (As a project from protocol to report) for SIP system.
- QACALuses Kaye validator (AVS and 2000) for Advanced Thermal validation as well as high-temperature dry bath for Pre and Post calibration.
- Kaye Validator -AVS model
- Kaye Validator-2000 model
THERMAL VALIDATION SERVICES
- QACAL provides Turnkey Advanced Thermal validation services (As a project from protocol to report) for critical process equipment’s like Autoclave / Steam Sterilizer, Dry Heat sterilizer, sterilizing tunnel, Freeze Dryer / Lyophiliser etc.
- QACAL uses Kaye validator (AVS and 2000) for Advanced Thermal validation as well as high-temperature dry bath for Pre and Post calibration.
- Kaye Validator -AVS model
- Kaye Validator-2000 model
- Customised report can be provided along with calibration traceability reports.

TEMPERATURE MAPPING SERVICES
- Why Perform GMP Temperature Mapping?
- Clause 3.19 of the PIC/S GMP guide states:
- “Storage areas should be designed or adapted to ensure good storage conditions. In particular, they should be clean and dry and maintained within acceptable temperature limits. Where special storage conditions are required (e.g. temperature, humidity) these should be provided, checked and monitored.”
- What will happen if…….
- -Your facility lost power over a weekend?
- Would you be set back months or even years in research and development?
- -Your refrigerator motor short circuits?
- Would you have to discard valuable material or otherwise sellable inventory?
- -You were faced with a surprise audit?
- Is your QA team ready? Do you readily have the documentation to avoid a deviation?

- Facility, warehouse, laboratory equipment and storage area temperature/RH mapping (validation) services
- QACAL provides Turnkey ((As a project from protocol to report) thermal validation/mapping in Entire facility which includes clean rooms, QC laboratory and warehouses as well as freezer rooms, cold rooms, temperature-controlled storage areas, quarantine areas and receiving and loading bays etc.
- Thermal validation/ mapping is performed as per WHO guidelines, PICS, Food and Drug Administration (FDA) or as per ICH.
- Temperature mapping/validation is performed using 21 CFR part -11 compliance software.
UTILITY VALIDATION
Compressed Air / Nitrogen Gas validation
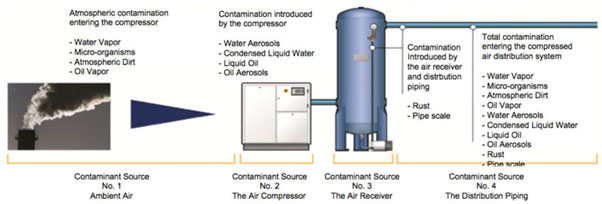
Compressed Air validation is a critical component in the production of pharmaceutical industry and effects on the quality of the end product. The Quality of Compressed air is important to ensure that product is safe. Compressed air validation is performed as per ISO 8573(Compressed Air – Contaminants and purity classes).
Without proper treatment, contaminants can travel from ambient air all the way through the compressed air system.
QACAL qualify Compressed air system with respect to contaminants of particles, water, oil, gases and microbiological contaminants.
The list of tests carried out under Compressed Air validation.
- Particle Count using HPD
- Microbial particle count
- Pressure Dew Point test
- Water vapour test (Moisture content)
- Odour Test
- Appearance test
- Carbon Dioxide
- Carbon Monoxide
- Nitrogen Monoxide and Nitrogen Dioxide
- Sulphur Dioxide
- Assay content of oxygen.
- Nitrogen purity test.
- Oil mist

- Steam Quality Test.
Steam quality testing is critical when performing any steam sterilization activity with an air removal step, such as autoclaving equipment.
International regulatory standards such as EN 285, EN 17665 & HTM2010 recommends steam quality testing and has defined parameters for compliance of clean steam. It is defined as the steam whose condensate meets all parameters of WFI. It must meet the basic physical properties as under.
QACAL performs Steam quality test using steam quality test kit.
FLOW PATTERN STUDY/SMOKE STUDY- TURNKEY SERVICE
Observations and solution
A review of Form 483’s issued to the 45-registered and inspected 503B Outsourcing Facilities identifies that over 40% (n = 19) of the inspected facilities received one or more observations concerning inadequate smoke studies.
Common findings of the smoke studies included studies that were not performed under dynamic conditions, studies that do not adequately reflect the aseptic processes or operator interventions performed, inadequate studies including limited smoke, studies that were not recorded, not reviewed, and final summary of acceptance, and studies where turbulent airflow was identified.
Detailed findings from 483s include the following:
- Dynamic Conditions, or not reflective of the firm’s processes:
- Smoke studies were not performed under dynamic conditions to verify that operators and processing equipment do not alter or impede the unidirectional cascade of air from the HEPA filters to the ISO 5 laminar flow benches where sterile drug products are opened and manipulated, and to the rest of the ISO 7 clean room.
- No smoke study procedure was used to evaluate your ISO 5 IV hood under dynamic conditions and no additional smoke studies have been conducted since the IV Hood was installed in 2009 [Note date of the inspection – August 2014].
- Smoke studies were not conducted for the ISO 5 hoods; they were only conducted in the ISO 7 cleanroom with [redacted] present, which is not representative of routine aseptic operations with [redacted] operators.
- A review of The Critical Manufacturing Area Smoke Test, completed 05/21/2014, found that the ISO 5 classified critical zones in each clean room were not evaluated under dynamic conditions with compounding equipment and components in place.
- Smoke studies are conducted during [redacted]; however, these studies are handled in static conditions and do not show adequate coverage of the ISO 5 area or the ISO 7/8 entryway and pass through(s).
- The firm’s in situ air pattern analysis (smoke studies) was not conducted under dynamic conditions, simulating routine production (i.e. compounding equipment in place and operations ongoing).
- Clean room certification and smoke studies for the ISO 7 Cleanroom were not performed during the operation of the [redacted].
- Smoke studies were not performed under dynamic conditions to verify that operators, processing equipment, or activities of the ISO 7 clean room do not alter or impede the unidirectionality of air from the HEPA filters to the [redacted] ISO 5 laminar flow benches where products are aseptically processes.
- Smoke studies are not performed under dynamic conditions.
- The smoke study was not performed under dynamic conditions to verify that the operator or activities in the ISO 7 cleanroom do not affect the unidirectional airflow from the HEPA filters in the ISO 5 hood where drug products are produced.
- Inadequate Studies:
- The smoke studies were not recorded to demonstrate laminar air flow during dynamic conditions.
- No dynamic airflow pattern studies (smoke studies) have been performed in the [redacted] ISO 5 hoods inside your ISO 7 room where sterile drug products are formulated and filled.
- Limited smoke was used and did not completely confirm uni-directional flow inside the filling line and all of its components.
- Not all operators’ interventions were included such as multiple interventions conducted at the same time or line set-up activities.
- Operators were observed standing directly in front of wall air returns but video failed to follow the smoke pattern at this area.
- Operators were observed performing very slow movements while adding stoppers and opening [redacted] doors – unlike current practice observed during routine operations at the same line on 11/17/14.
- The smoke studies are not reviewed by your personnel and there is no final report regarding the adequacy of the airflow.
- The firm has no documentation of smoke studies conducted under dynamic conditions to indicate adequate unidirectional air control during sterile compounding.
- Smoke studies do not indicate vertical laminarity of air flow from your ISO 5 vertical flow hood.
- The smoke studies performed in the ISO 5 [redacted] area of SVP Line 1 are inadequate in that there is insufficient smoke to clearly show unidirectional air flow, the dynamic portion of the smoke study at the [redacted] included an operator who did not move, and the study did not include smoke near the window on the back side of holes that were open to the ISO 8 room in order to perform [redacted].
- Turbulent Air Flow:
- Smoke studies conducted on March 20111 2014 show turbulent and stagnant air within ISO 5 areas used to sterilize and fill drug product unit containers.
- Video of the dynamic smoke studies conducted to demonstrate unidirectional airflow [redacted] located in clean room [redacted] showed considerable turbulent airflow when [redacted] was placed within the work zones of the [redacted] l
QACAL has significant experience in the planning, development, and execution of critical airflow visualization studies commonly referred to as Smoke Studies.
We provide solution of all the above problem with expertise in aseptic activity capturing and data processing.
Video Recording- Our Expertise in Processing
Video recording of a Smoke study is a very useful aide in investigating personnel behaviour that could negatively affect the aseptic manufacturing process.
QACAL uses advance multi angle camera system to perform videography without human intervention.
We provide multi angle simulation video as final proof of smoke study Qualification. For generating multi angle simulation video, we use high end software.
We can visualize the videography recording remotely during smoke study.
MEDIA FILL-PROTOCOL TO VIDEOGRAPHY
Media fill:
Media fill is a key qualification of an aseptic facility.
A media fill (Also known as a process simulation) is the performance of an aseptic manufacturing procedure using a sterile microbiological growth medium in place of the drug solution.
We develop complete media fill protocol, executions, analysis of results and conclusion independently.
Our protocol includes
- Factors associated with the longest permitted run on the processing line that can pose contamination risk (e.g. operator fatigue)
- Representative number, type, and complexity of normal interventions that occur with each run, as well as non-routine interventions and events (e.g. maintenance, stoppages, equipment adjustments)
- Define allowable interventions
- Define grades of interventions
- Practise all interventions during media runs
- Lyophilisation, when applicable
- Aseptic assembly of equipment (e.g. at start-up, during processing)
- Number of personnel and their activities
- ‘worst case’ conditions, which typically involve the largest container with the widest mouth or small ampoules running at high speed with frequent jamming.
- Timings, Days, Line speed, Size of container, number of containers


Video recording of a media fill test validation maybe a useful aide in
investigating personnel behavior that could negatively affect the aseptic
manufacturing process.
Video Recording- Our Expertise in Processing
Video recording of a media fill validation may be a useful aide in investigating personnel behaviour that could negatively affect the aseptic manufacturing process.